
いま我々がお世話になっている医薬品もかつては新薬だった――。さまざまな病気の予防や治療、症状の緩和や改善をめざして、これからも日夜研究・開発が続けられ、臨床試験などを経たのち新薬となる、創薬には膨大な時間と費用を要する。
身体の仕組みや病気・症状について新しい知識が得られると、新薬候補物質の探索や合成が試みられる。創薬の初期段階においてさえそれら候補化合物が新薬になる率は1/10,837(参照:製薬協Webサイト)。
そうしたなか世界的に、コンピュータを利用した新薬開発手法(IT創薬)に高い関心が寄せられている。IT創薬は、実際に化合物の生成と実験を繰り返すのではなく、化学物質を仮想的に設計し、効果を見積もれる画期的な技術として期待されている。しかしこれまで、量子力学に基づく手法は年単位の計算時間を要するため使えず、よく用いられる技法はその精度に課題があったという。
富士通研究所は、薬効の目安すなわち疾病の原因となるタンパク質(標的タンパク質)の形状に合わせて化学物質がその形状を変化させる度合い、標的タンパク質と薬の候補物質が引き合う強さである結合強度を、精度よく推定できる分子シミュレーション技術を開発した。
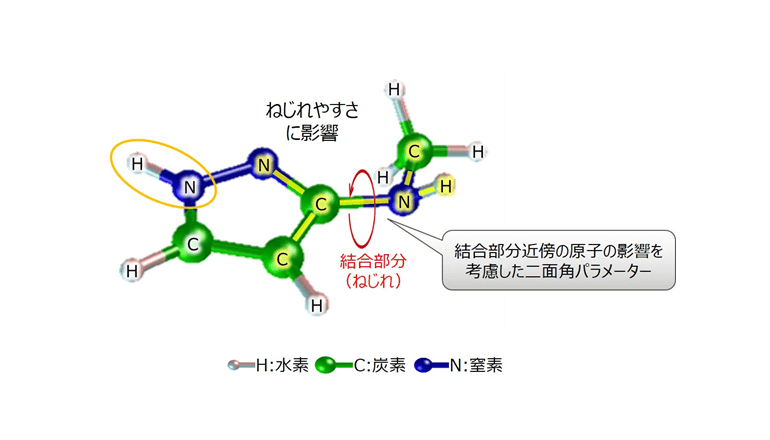
結合強度の予測では従来、高速かつ標的タンパク質のような大きな分子も容易に取り扱えるため、分子内の原子間に働く力をニュートン力学により近似的に算出する分子シミュレーション技術が広く用いられてきたが、これは最重要パラメーターである結合部分のねじれ度合いの推定精度が低いため、結合強度の推定精度も低くなっていたという。
同社の分子シミュレーション技術は、ねじれ度合いについて、結合部分だけでなくその近傍の原子の影響まで考慮して推定。190種類の化学物質で、第一原理計算による正しい結果との誤差を評価したところ、従来技術(GAFF1.8)に比べて推定誤差が平均1/10以下だったとのことだ。